
- Accueil >
- Pathologies >
- Malformations faciales >
- Syndrome de Sturge Weber
Syndrome de Sturge-Weber
Le syndrome de Sturge-Weber est une maladie rare touchant la peau, l'oeil et le système nerveux (maladie oculo-neuro-cutanée) et présente à la naissance (congénitale).
Il est caractérisé par des malformations des vaisseaux sanguins (angiomes) au niveau :
- du visage, à l'origine d'une tache de naissance (angiome plan) ;
- du cerveau et des yeux, à l'origine de troubles neurologiques et visuels.
Syndrome de Sturge-Weber
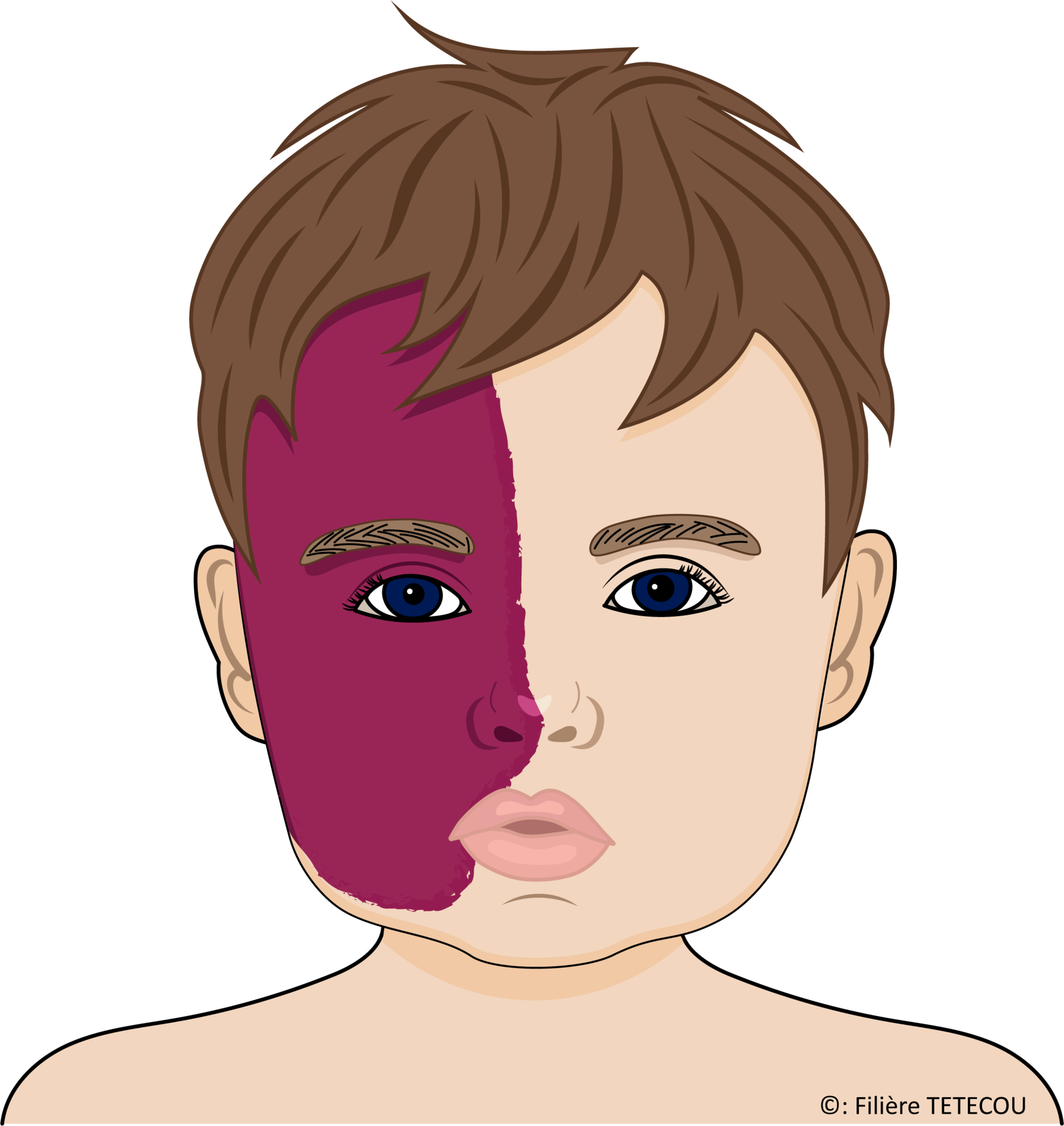
On distingue généralement 3 types de syndrome de Sturge-Weber selon les organes affectés :
- le type I (syndrome de Sturge-Weber classique) : les angiomes concernent le visage et la surface du cerveau. L'épilepsie et le glaucome sont fréquents.
- le type II : les angiomes touchent le visage mais pas le cerveau. Un glaucome peut survenir.
- le type III : les angiomes n'affectent que la surface du cerveau.
La prévalence à la naissance en Europe est estimée entre 1/20 000 et 1/50 000.
La cause
Le syndrome de Sturge-Weber est dû à la mutation du gène GNAQ, localisé sur le chromosome 9 (9q21.2), ou du gène GNA11, sur le chromosome 19 (19p13.3). Une mutation du gène GNB2 , sur le chromosome 7 (7q22.1), a été décrite chez un patient.
Cette nouvelle mutation dite spontanée n'a pas été transmise par les parents mais s'est produite accidentellement dans certaines cellules du fœtus, au cours de son développement : on parle de mutation somatique en mosaïque. Certaines cellules sont porteuses de la mutation et d'autres non.
Comme la mutation n'est pas présente dans les cellules germinales, ovocytes et spermatozoïdes, elle n'est pas transmise à la génération suivante.
La mutation génétique provoque des anomalies des vaisseaux sanguins et par conséquent une mauvaise circulation sanguine dans la zone de distribution d'une ou plusieurs des trois branches du nerf facial (5e nerf crânien ou nerf trijumeau) pouvant s'étendre jusqu'à la surface du cerveau sous-jacente.
Les yeux peuvent également être affectés par ces anomalies vasculaires.
Les mutations du gène GNAQ peuvent aussi être à l'origine d'une tache de naissance sans troubles visuel ou neurologique associés.
Les manifestations
Le syndrome de Sturge-Weber est à l'origine de plusieurs manifestations dont la présence et la sévérité varient d'une personne à l'autre.
-
-
Angiome cutané
-
Cette tache de naissance sur la peau du visage est de couleur rouge ou pourpre et se compose de vaisseaux sanguins minces et malformés.
L'étendue de l'angiome correspond à toute ou une partie de la zone de répartition du nerf facial (5e nerf crânien ou nerf trijumeau) avec ses trois branches.
L'angiome couvre habituellement le front et la paupière supérieure d'un côté du visage.
Chez une personne sur cinq, il touche les deux côtés du visage.
Contrairement aux angiomes cutanés isolés, cette tache de naissance n'a pas tendance à régresser avec le temps.
Cet angiome peut avoir un impact esthétique important avec des répercussions sur l'estime de soi et le regard des autres, susceptibles de contrarier l'inclusion sociale, scolaire et professionnelle de la personne atteinte.
 Crédit : Licence Creative Commons, adapté de BruceBlaus par Filière TETECOU
Crédit : Licence Creative Commons, adapté de BruceBlaus par Filière TETECOU
-
-
Troubles visuels
-
La présence de malformations vasculaires dans l'œil (angiome choroïdien) est à l'origine d'un glaucome qui en est la complication oculaire la plus fréquente, avec 30 à 60 % des personnes concernées.
Près de la moitié des personnes atteintes développent un glaucome généralement dans la petite enfance mais il peut également apparaître plus tard dans l'enfance ou à l'âge adulte.
Le glaucome correspond à une pression de liquide anormalement élevée dans l'œil se manifestant par une intolérance à la lumière (photophobie), une augmentation des larmes (flux lacrymal), une opacification de la cornée, une hypertrophie de l'œil (buphtalmie) et une déficience visuelle.
Les malformations vasculaires présentent au niveau du cortex visuel (la zone la plus en arrière du cerveau) provoquent une perte du champ visuel unilatéral (hémianopsie).
- Troubles neurologiques
Les malformations vasculaires à la surface du cerveau sont à l'origine d'une épilepsie, c'est à dire un dérèglement soudain et transitoire de l'activité électrique du cerveau se manifestant par la survenue de crises d'épilepsie (absences, spasmes, convulsions...), chez 70 à 90 % des personnes atteintes.
L'épilepsie peut apparaître à tout moment jusqu'à l'âge de trois ans, et près de 75 % des enfants en sont atteints au cours de leur première année de vie.
Plus l'épilepsie débute tôt dans la vie, plus le risque de paralysie de la moitié du corps (hémiparésie) et l'impact sur le développement cognitif sont importants avec une déficience intellectuelle ou d'autres problèmes d'apprentissage, de mémoire ou de comportement concernant près de 50 à 75 % des personnes atteintes.
Il peut également se produire, plus tard dans la vie, des épisodes transitoires d'altération de la circulation sanguine dans le cerveau, appelés attaques ischémiques transitoires (AIT) caractérisées par des manifestations semblables à celles d'un accident vasculaire cérébral (AVC) mais de courte durée (baisse de force ou de sensation d'un côté du corps, déformation du visage, trouble de la parole, trouble de la vision...).
-
Autres manifestations
En fonction de sa localisation, l'angiome facial peut être à l'origine, à l'âge adulte, d'une croissance excessive (hypertophie) des tissus mous de la bouche et du visage, ou de l'os de la mâchoire inférieure (mandibule) à l'origine de troubles d'élocution, de la mastication, d'anomalies dentaires.
Des anomalies vasculaires peuvent également être présentes sur d'autres parties de la peau ou dans les organes internes.
Il existe un déficit moteur chez près de la moitié des personnes atteintes.
De faibles niveaux d'hormone thyroïdienne et d'hormone de croissance ont été décrits dans ce syndrome.
Le diagnostic
Le diagnostic du syndrome de Sturge-Weber repose sur l'observation de la tache de naissance caractéristique sur le visage du nourrisson.
Le diagnostic peut être confirmé par l'imagerie par résonance magnétique (IRM) cérébrale ou par une tomodensitométrie (TDM).
L'électroencéphalographie (EEG) permet de diagnostiquer et localiser l'épilepsie.
Le syndrome de Sturge-Weber doit être différencié d'un angiome facial isolé et des syndromes d'hypercroissance liés au gène PIK3CA, en particulier le syndrome mégalencéphalie-malformation capillaire-polymicrogyrie (MCAP).
La prise en charge
La prise en charge globale de la personne atteinte du syndrome de Sturge-Weber et de sa famille repose sur une coopération pluridisciplinaire médicale pouvant impliquer, selon les manifestations : pédiatre, néonatologue, dermatologue, neuropédiatre, neurologue, neurochirurgien, neuroradiologue, dermatologue, ophtalmologiste, pédopsychiatre, endocrinologue, dentiste, orthodontiste, chirurgien maxillo-facial, chirurgien plastique, etc.,
ainsi qu'une équipe de professionnels paramédicaux et sociaux : orthoptiste, infirmier, orthophoniste, kinésithérapeute, psychomotricien, ergothérapeute, orthoprothésiste, psychologue, éducateur spécialisé, assistant social, auxiliaire de vie sociale,
dont la coordination peut être assurée par un des spécialistes, en lien avec le médecin traitant.
Au sein de la Filière TETECOU, les personnes atteintes du syndrome de Sturge-Weber peuvent être prises en charge dans :
-
les Centres de Référence ou de Compétence des Fentes et Malformations faciales (MAFACE) pour la prise en charge de l'angiome du visage et d'autres malformations faciales,
-
les Centres de Référence ou de Compétence des Syndromes de Pierre Robin et Troubles de succion-déglutition congénitaux (SPRATON)
-
les Centres de Référence ou de Compétence des Maladies Rares Orales et Dentaires (O-Rares) pour leurs soins dentaires,
-
les Centres de Référence ou de Compétence des Malformations ORL Rares (MALO) pour la prise en charge des malformations ORL.
 |
 |
-
Prise en charge de l'angiome facial
Le traitement au laser à colorant pulsé peut être proposé dès l'enfance pour réduire cette tache de naissance.
Des conseils de maquillage peuvent également être donnés aux personnes concernées.
- Prise en charge neurologique
L'épilepsie nécessite un traitement médicamenteux anti-épileptique et, dans certains cas sévères, une intervention neurochirurgicale peut être proposée.
- Prise en charge ophtalmologique
Une surveillance ophtalmologique précoce et régulière est nécessaire tout au long de la vie.
Un traitement par des collyres permet de diminuer la pression intraoculaire et sera complété par des interventions chirurgicales selon les cas.
-
Prise en charge maxillo-faciale
Des interventions chirurgicales peuvent être nécessaires pour réduire le volume de la lèvre inférieure, lorsque l'angiome facial est à l'origine de son hypertrophie (croissance excessive).
Une chirurgie orthognathique peut êre proposée en cas d'hypertrophie ou de croissance asymétrique de la mandibule, l'os de la mâchoire inférieure.
-
Prise en charge bucco-dentaire
Lorsque l'angiome facial couvre la région de la bouche et du menton, la gencive est fréquemment atteinte. Elle est alors épaissie avec un degré de sévérité variable ; cela peut être très léger ou gêner la fermeture de la bouche. Cette hyperplasie gingivale, si elle est importante, peut nécessiter une intervention chirurgicale.
-
Prise en charge des troubles hormonaux
Des traitements spécifiques peuvent corriger les taux insuffisants d'hormones thyroïdiennes et de croissance.
-
Prise en charge des troubles du langage et de la communication
Si le langage est retardé et qu'il y a des difficultés d'élocution, une prise en charge orthophonique est indispensable.
Les parents et l'entourage reçoivent des conseils sur la façon dont le langage peut être stimulé. Si nécessaire, ils sont également accompagnés dans l'utilisation de moyens de communication alternatifs pour donner à l'enfant les conditions pour comprendre et s'exprimer en fonction de ses capacités.
L'enseignement doit être adapté au profil cognitif et aux capacités de perception de l'enfant.
- Prise en charge psychologique
La famille doit bénéficier d'un soutien psychologique au moment du diagnostic et également plus tard. Les enfants et les jeunes eux-mêmes devraient également, si nécessaire, bénéficier d'un soutien continu par un psychologue en fonction de leur âge et de leur maturité.
L'Education Thérapeutique du Patient (ETP)
L'Education Thérapeutique du Patient (ETP) fait partie intégrante du parcours de prise en charge. Ces interventions éducatives, centrées sur la personne malade, lui permettent de mieux vivre au quotidien, de prévenir les complications et d'être actrice de sa santé.

Les personnes atteintes du syndrome de Sturge-Weber peuvent bénéficier de programmes d'Éducation Thérapeutique du Patient au sein de la Filière TETECOU :
![]() Hôpital Necker-Enfants malades, Paris
Hôpital Necker-Enfants malades, Paris

Éducation thérapeutique pour la prévention des complications et l'amélioration de la qualité de vie des enfants atteints de malformations de la face et de la cavité buccale
![]() Hôpital Necker-Enfants malades, Paris
Hôpital Necker-Enfants malades, Paris
![]()
E...change de regard : Programme destiné aux enfants (entre 6 et 11 ans) ayant une maladie rare et souffrant du regard des autres
![]() Hôpitaux Universitaires de Strasbourg
Hôpitaux Universitaires de Strasbourg

DentO-RarEduc : Programme destiné aux patients souffrant de maladies rares des dents et/ou de la cavité buccale
En fonction du centre de prise en charge, d'autres programmes d'Education Thérapeutique du Patient peuvent être proposés, tels que
![]() CHU de Bordeaux
CHU de Bordeaux
![]()
Éducation thérapeutique des patients porteurs d'une maladie rare : TEMARA
![]() CHU de Bordeaux
CHU de Bordeaux
![]()
CAP’ADJA pour l’adolescent et le jeune adulte porteur de maladie rare
![]() CHU de Clermont-Ferrand
CHU de Clermont-Ferrand
![]()
Coup de pouce vers l'avenir : Éducation thérapeutique pour les patients suivis dans le service de génétique médicale
![]() CHU de Dijon
CHU de Dijon
![]() CHU de Lille
CHU de Lille
![]() Hospices Civils de Lyon
Hospices Civils de Lyon
![]()
E...change de regard : Programme destiné aux enfants (entre 6 et 11 ans) ayant une maladie rare et souffrant du regard des autres
Des ateliers éducatifs, sur l'estime de soi, par exemple, ou des rencontres avec d'autres parents ou enfants peuvent également être organisés.

Ressources
Associations de personnes malades
Association
Syndrome Sturge-Weber France
![]()
Association
Anna
![]()
Orphanet et sites d'intérêt


Documentation et livrets d'information pour les personnes malades et leurs familles



Recommandations et autres références pour les professionnels
Protocole National de Diagnostic et de Soins
Février 2024
avec la collaboration de :
- CRMR des Maladies Rares Orales et Dentaires




Ressources médico-sociales



Médias


"Des outils numériques pour informer les professionnels et les familles"
par Mme Alexandra Richer, Association Vanille-Fraise,
4e Journée nationale de la filière TETECOU, Rencontre des associations, 4 octobre 2019
Vidéo de We are patients
wearepatients.com
Témoignage de François-Régis, de l'Association Vanille-Fraise