![]()
Les RASopathies
Le terme "RASopathies" désigne un ensemble de syndromes génétiques qui ont en commun le fait d'être lié à une dérégulation (augmentation/diminution) des gènes impliqués dans la voie de signalisation RAS/MAPK. Cette voie de signalisation joue un rôle essentiel dans le contrôle de la prolifération, de la différenciation (spécialisation d'une cellule dans un "type cellulaire"), de l'apoptose (mort cellulaire programmée) et de la migration des cellules. De ce fait, cette voie est impliquée dans le développement de l'embryon et dans la cancérogenèse.
Parmis les RASopathies on retrouve la neurofibromatose de type 1, le syndrome de Noonan, le syndrome cardio-facio-cutané, le syndrome de Costello, le syndrome de Noonan avec lentigines multiples, le syndrome de Legius, etc.
Les pathologies qui nécessitent une prise en charge au sein d'un centre spécialisé SPRATON (du fait de la forte prévalence de difficultés alimentaires) sont principalement : le syndrome de Noonan, le syndrome cardio-facio-cutané et le syndrome de Costello.
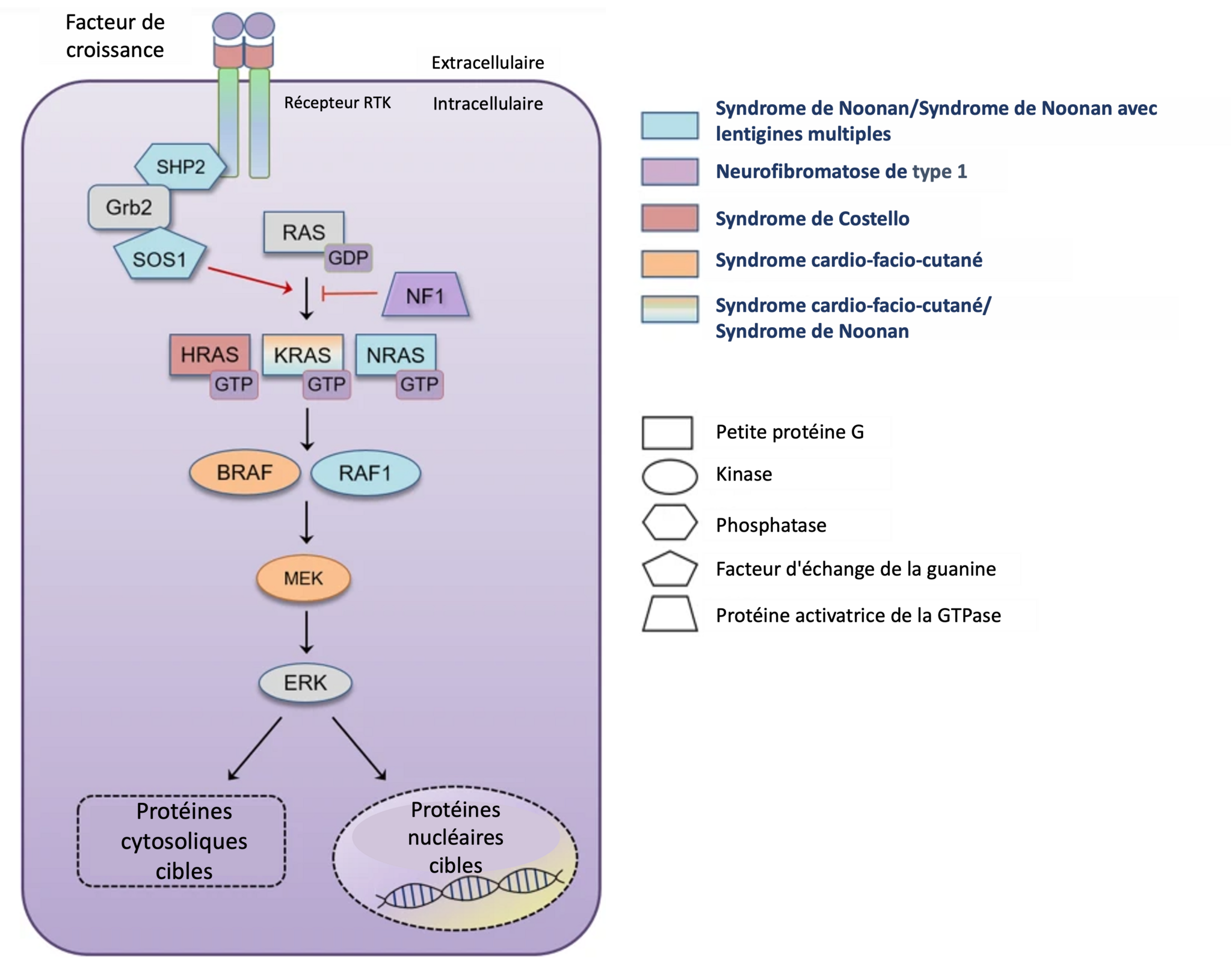
Voie RAS-MAPK et Rasopathies.
Schéma simplifiée de la voie de signalisation RAS-MAPK annoté par un code couleur indiquant les gènes mutés dans les principales RASopathies. Chaque couleur correspond à une (ou deux) RASopathie(s) spécifique(s). Cette représentation n'est pas exhaustive de toutes les altérations génétiques identifiées. Les gènes sont également représentés sous forme de polygone selon leurs catégories fonctionnelles.
Explication simplifiée de la voie de signalisation : Un signal extérieur se fixe au récepteur tyrosine kinase à la surface de la cellule (réception du signal extracellulaire). L’activation de ce récepteur entraîne l’activation de la protéine RAS, qui agit comme un interrupteur moléculaire (activation de RAS). Ce processus déclenche une série d'activités enzymatiques connue sous le nom de cascade MAPK (activation de la cascade MAPK). Le message atteint le noyau (transduction du signal), ce qui entraîne une réponse adaptée de la cellule (réponse cellulaire).
Crédit : Adaptation de Kang, M., Lee, YS. The impact of RASopathy-associated mutations on CNS development in mice and humans. Mol Brain 12, 96 (2019). https://doi.org/10.1186/s13041-019-0517-5). Licence CC-BY 4.0/2.0.
Le syndrome de Noonan est la plus commune des RASopathies.
Sa prévalence à la naissance est de l'ordre de 1 naissance sur 1000 à 1 naissance sur 2500.
Ce syndrome touche indifféremment les garçons et les filles.
Causes et diagnostic
Le diagnostic peut être clinique, basé sur l'observation des manifestations caractéristiques du syndrome de Noonan (traits du visage, problème cardiaque, petite taille…).
La confirmation du diagnostic est possible dans près de la moitié des cas par une simple analyse de sang.
Une anomalie génétique est identifiée chez plus de 80% des patients. Les gènes les plus fréquemment mutés étant le gène PTPN11 (ou SHP2) (chez environ 50 % des patients), suivi du gène SOS1 (chez 10 à 15 % des patients), RAF1 (chez 5 % des patients), RIT1 (chez 5 % des patients) ou encore KRAS (chez moins de 5 % des patients). On retrouve les gènes BRAF, LZTR1, MAP2K1 et NRAS mutés chez moins d'1 % des patients. Tous ces gènes codent pour des constituants ou des régulateurs de la voie de signalisation RAS/MAPK.
Le syndrome de Noonan est une maladie génétique qui se transmet de manière autosomique dominante. Une personne atteinte a donc une probabilité de 50% d'avoir des enfants également touchés par ce syndrome, indépendamment de leur sexe et du gène impliqué.
Dans la plupart des cas, la mutation est dite de novo (non héritée des parents) mais il est également possible qu'elle soit transmise par l'un des deux parents.
Manifestations
Les manifestations et leur sévérité sont variables entre les personnes atteintes, et évoluent au cours du temps.
Parmi les manifestations possibles, on retrouve :
-
Une malformation cardiaque dans 80% des cas : caractérisée fréquemment par une sténose des valves pulmonaires (rétrécissement des valves de l'artère pulmonaire) et/ou une cardiomyopathie hypertrophique (épaississement du muscle cardiaque);
-
Des difficultés alimentaires avec notamment faible appétit, trouble de succion, refus alimentaire et un reflux gastro-oesophagien.
-
Des particularités morphologiques faciales qui peuvent comprendre : un hypertélorisme (yeux écartés), un ptosis (affaissement de la paupière supérieure souvent unilatérale), des fentes palpébrales dirigées vers le bas et le dehors, un cou court et large, oreilles basses, épaisses et basculées en arrière... ;
-
Des difficultés d'apprentissage et rarement une déficience intellectuelle ;
-
Des anomalies buccodentaires telles que des malocclusions des arcades dentaires et/ou hypo-développement mandibulaire. Des anomalies de nombre, de la forme ou de l'éruption des dents peuvent être observées ;
-
Des troubles de la vision et de l'audition ;
-
Des anomalies modérées dans le processus de coagulation du sang ;
Certaines recherches ont exploré le lien entre les gènes altérés et la sévérité des manifestations cliniques. Par exemple il a été montré que les mutations du gène SOS1 sont généralement liées à des formes plus légères du syndrome de Noonan. Elles sont moins souvent associées à un déficit statural (taille inférieure à la moyenne) ou à une déficience intellectuelle. En revanche, les anomalies ectodermiques telles que des cheveux frisés et une kératose pilaire sont plus courantes chez ces patients. A l'inverse, les individus porteurs de mutations du gène PTPN11 présentent plus fréquemment un déficit statural, une déficience intellectuelle, des anomalies squelettiques ainsi que des traits morphologiques faciaux plus marqués (Roberts et al. 2013 ; Reynolds et al. 2025).
En ce qui concerne les troubles de l'alimentation (digestion, appétit), les données actuelles ne montrent pas de distinction spécifique au niveau des gènes.
La prise en charge
Prise en charge au sein du réseau SPRATON
Aider à l'alimentation
Chez les nouveau-nés et les nourrissons atteints d’un syndrome de Noonan, les difficultés d'alimentation sont très fréquentes (plus de 2/3 des cas) : certains enfants qui présentent une hypotonie, c'est-à-dire un tonus musculaire faible, rencontrent des problèmes pour téter (succion faible) diminuant ainsi leur apport calorique.
Ces difficultés d'alimentation se traduisent par des anomalies de la croissance pondérale et durant les premiers mois de vie, leur poids descend en dessous de la norme.
Pour aider à une alimentation suffisante, les enfants pourront avoir besoin d'une assistance durant les premiers mois de vie :
- soit par des conseils diététiques pour enrichir l'alimentation et faciliter la succion
- soit une nutrition entérale de soutien transitoire à l’aide d’une sonde introduite par le nez et qui va dans l’estomac (sonde naso-gastrique) ; soit par une sonde qui délivrera les aliments directement dans l’estomac, après une intervention appelée gastrostomie.
Généralement, après deux ans, ces difficultés alimentaires s'estompent.
Le reflux gastro-œsophagien (RGO) peut être fréquent chez les enfants porteurs du syndrome de Noonan. Pour traiter le RGO, du lait AR anti régurgitations peut être utilisé ainsi que des traitements inhibiteurs de la pompe à protons ou anti-H2.
Retrouvez des conseils, des infos et des outils sur les difficultés alimentaires de votre enfant sur le site ![]()
Prise en charge au sein des autres réseaux
Compte tenu des nombreuses manifestations cliniques, la prise en charge globale des patients atteints du syndrome de Noonan nécessite une coopération pluridisciplinaire médicale, paramédicale et sociale. Pour la prise en charge des troubles ORL, des malformations faciales et des soins dentaires, le patient pourra être pris en charge au sein des autres réseaux TETECOU.
En savoir plus sur la prise en charge au sein des autres réseaux
Pour les atteintes intellectuelles, cardiaques, squelettiques (etc.) la prise en charge peut se faire au sein des filières santé maladies rares : Défiscience, AnDDI-Rares, Oscar et Cardiogen.
Pour aller plus loin
Association de patients
Association Noonan
![]()
Ressources Orphanet


Recommandations pour les professionnels
Protocole National de Diagnostic et de Soins
RASopathies : syndromes de Noonan, cardio-facio-cutané et apparentés


Ressources TETECOU
Podcast TETECOU

Le syndrome cardio-facio-cutané (CFC) est une RASopathie dont la prévalence à la naissance est de moins d'1/20 000.
Le syndrome CFC est morphologiquement proche du syndrome de Noonan (RASopathie la plus commune) et présente le même spectre de complications bien qu'il soit plus marqué, en particulier sur le plan neurocognitif avec l'existence presque permanente d'un retard psychomoteur et/ou d'une déficience intellectuelle, fréquemment sévère.

Illustration du visage, vu de face, d'un enfant atteint du syndrome cardio-facio-cutané
Causes et diagnostic
Le diagnostic clinique est évoqué sur la base de l'observation des manifestations caractéristiques du syndrome cardio-facio-cutané (traits du visage, dermatoses, petite taille…) mais doit être confirmé par analyse génétique. En effet, dans plus de 80% des cas, un test génétique permet de confirmer le diagnostic clinique et les patients sans mutation identifiable sont rares.
Dans le cas du syndrome CFC, les mutations génétiques sont quasiment toujours sporadiques et surviennent généralement de novo (elles ne sont pas héritées des parents). Les mutations en question touchent particulièrement les gènes BRAF (dans 75% des cas), KRAS, MAP2K1 et MAP2K2 de la voie de signalisation RAS/MAPK.
Manifestations
Les manifestations et leur sévérité sont variables entre les personnes atteintes, et évoluent au cours du temps.
Parmi les manifestations possibles, on retrouve :
-
Une malformation cardiaque dans 80% des cas : caractérisée fréquemment par une cardiomyopathie hypertrophique (épaississement du muscle cardiaque) dans 70% des cas et/ou une sténose des valves pulmonaires (rétrécissement des valves de l'artère pulmonaire) ;
- Des difficultés alimentaires dans plus de 70% des cas avec notamment faible appétit, refus alimentaire, troubles de la déglutition, troubles de la succion et reflux gastro-oesophagien.
-
Des particularités morphologiques faciales similaires au syndrome de Noonan bien que plus marquées : un hypertélorisme (yeux écartés), un ptosis (affaissement de la paupière supérieure), des fentes palpébrales dirigées vers le bas et le dehors, un cou court et large, oreilles basses, épaisses et basculées en arrière... ;
- Des dermatoses (affections de la peau) dont les manifestations peuvent être : des cheveux clairsemés, fins et bouclés, une peau sèche, hyperkératosique et hyperélastique, des nævi (tâches pigmentées), une ichtyose (sécheresse), un lymphoedème, des hémangiomes...;
- Des troubles neurologiques qui peuvent comprendre : une hypotonie (baisse de la tonicité musculaire), un retard de langage, une macrocéphalie (augmentation du volume de la tête), une épilepsie, une déficience intellectuelle... ;
- Des troubles de la vision et de l'audition ;
-
Des anomalies buccodentaires ;
- Des anomalies musculosquelettiques ;
La prise en charge
Prise en charge au sein du réseau SPRATON
Aider à l'alimentation
Les difficultés alimentaires sont fréquentes chez les nourrissons atteints du syndrome CFC et entrainent un retard de croissance. Elles nécessitent donc une prise en charge spécialisée.
Certains enfants qui présentent une hypotonie, c'est-à-dire un tonus musculaire faible, rencontrent des problèmes pour téter (succion faible) diminuant ainsi leur apport calorique.
Pour aider à une alimentation suffisante, les enfants pourront avoir besoin d'une assistance durant les premiers mois de vie :
- soit par des conseils diététiques pour enrichir l'alimentation et faciliter la succion
- soit une nutrition entérale de soutien transitoire à l’aide d’une sonde introduite par le nez et qui va dans l’estomac (sonde naso-gastrique) ; soit par une sonde qui délivrera les aliments directement dans l’estomac, après une intervention appelée gastrostomie.
Généralement, après deux ans, ces difficultés alimentaires s'estompent.
Le reflux gastro-œsophagien (RGO) peut être fréquent chez les enfants porteurs du syndrome de Noonan. Pour traiter le RGO, du lait AR anti régurgitations peut être utilisé ainsi que des traitements inhibiteurs de la pompe à protons ou anti-H2.
Il pourra être nécessaire de suivre des séances orthophoniques de rééducation et de préservation de l'oralité.
L'orthophoniste, formé à la prise en charge des troubles alimentaires, est en mesure de recommander les gestes, les équipements et les postures appropriés pour faciliter l'alimentation
Une prise en charge orthophonique pour le retard de langage et de parole est également indispensable.
Retrouvez des conseils, des infos et des outils sur les difficultés alimentaires de votre enfant sur le site ![]()
Prise en charge au sein des autres réseaux
Compte tenu des nombreuses manifestations cliniques, la prise en charge globale des patients atteints du syndrome CFC nécessite une coopération pluridisciplinaire médicale, paramédicale et sociale. Pour la prise en charge des troubles ORL, des malformations faciales et des soins dentaires, le patient pourra être pris en charge au sein des autres réseaux TETECOU.
En savoir plus sur la prise en charge au sein des autres réseaux
Pour les atteintes intellectuelles, cardiaques, squelettiques (etc.) la prise en charge peut se faire au sein des filières santé maladies rares : Défiscience, AnDDI-Rares, Oscar et Cardiogen.
Pour aller plus loin
Association de patients
Association du syndrome de Costello et CFC
![]()
Recommandations pour les professionnels
Protocole National de Diagnostic et de Soins
RASopathies : syndromes de Noonan, cardio-facio-cutané et apparentés
Ressources Oprhanet

Ressources TETECOU
Page TETECOU
Syndrome CFC

Podcast TETECOU
Le syndrome de Costello est une des RASopathies les plus rares dont la prévalence est estimée entre 1 naissance sur 300 000 et 1 naissance sur 1 250 000.
Ce syndrome touche indifféremment les garçons et les filles.
Causes et diagnostic
Le diagnostic clinique est évoqué sur la base de l'observation des manifestations caractéristiques du syndrome de Costello (comportement neurologique néonatal (irritabilité, hypotonie...), caractéristiques de la peau, problèmes cardiaques, apparence du visage...). Néanmoins, un test génétique peut confirmer le diagnostic.
Dans le cas du syndrome de Costello, les mutations génétiques surviennent généralement de novo (elles ne sont pas héritées des parents). Les mutations en question touchent exclusivement le gène HRAS impliqué dans la voie de signalisation RAS/MAPK.
Manifestations
Les manifestations et leur sévérité sont variables entre les personnes atteintes, et évoluent au cours du temps.
Parmi les manifestations possibles, on retrouve :
-
Une malformation cardiaque caractérisée par une sténose des valves pulmonaires (rétrécissement des valves de l'artère pulmonaire) et/ou une cardiomyopathie hypertrophique (épaississement du muscle cardiaque) et/ou des arythmies (troubles du rythme cardiaque) ;
-
Un retard statural et des difficultés alimentaires avec notamment des troubles de la déglutition, constipation, dégoût alimentaire (jusque 3-4 ans) et un reflux gastro-oesophagien ;
-
Des traits du visage particulièrement marqués qui peuvent comprendre : un hypertélorisme (yeux écartés), un ptosis (affaissement de la paupière supérieure), des oreilles basses, épaisses et basculées en arrière, des lèvres charnues, un crâne large (macrocéphalie), des cheveux peu fournis et frisés...;
-
Des difficultés d'apprentissage et un retard intellectuel léger à modéré ;
-
Une hypersensibilité sensorielle et une hyperémotivité très marquée, avec parfois des troubles du spectre autistique ;
-
Des troubles ORL et de la vision ;
-
Des anomalies buccodentaires dont des gencives trop épaisses (hypertrophie gingivale), des malocclusions dentaires... ;
-
Des anomalies musculo-squelettiques telles qu'une scoliose, des anomalies de position des pieds... ;
-
Des anomalies de la peau qui peuvent comprendre : des papillomes (excroissance de peau ressemblant à des verrues), une peau lâche, souple d'aspect trop abondante, une sudation excessive, un prurit (démangeaisons), des plis des paumes et des plantes des pieds profonds... ;
-
Un risque accru à développer certains cancers (10 à 15% de risque) justifiant une surveillance échographique semestrielle pendant les premières années de vie.
La prise en charge
Prise en charge au sein du réseau SPRATON
Aider à l'alimentation
Les difficultés alimentaires sont fréquentes chez les nourrissons atteints du syndrome de Costello et entrainent un retard de croissance. Certains enfants ont du mal à téter et à avaler et cela est probablement lié à un trouble du contrôle de la déglutition par le cerveau.
Ces difficultés d'alimentation nécessitent donc une prise en charge spécialisée.
Pour aider à une alimentation suffisante, les enfants auront fréquemment besoin d'une assistance dès les premiers mois de vie par nutrition entérale :
- soit à l’aide d’une sonde introduite par le nez et qui va dans l’estomac (sonde naso-gastrique)
- soit par une sonde qui délivrera les aliments directement dans l’estomac, après une intervention appelée gastrostomie.
Généralement, ces difficultés alimentaires s'estompent après l'âge de 4-5 ans.
Les enfants atteints du syndrome de Costello peuvent souffrir de reflux gastro-œsophagien (RGO). Pour traiter le RGO, du lait AR anti régurgitations peut être utilisé ainsi que des traitements inhibiteurs de la pompe à protons ou anti-H2. Une intervention chirurgicale anti-reflux de type Nissen pour un RGO sévère peut également être réalisée.
Il pourra être nécessaire de suivre des séances orthophoniques de rééducation et de préservation de l'oralité.
L'orthophoniste, formé à la prise en charge des troubles alimentaires, est en mesure de recommander les gestes, les équipements et les postures appropriés pour faciliter l'alimentation
Une prise en charge orthophonique pour le retard de langage et de parole est également indispensable. En effet, dans le cadre du syndrome de Costello, l’âge moyen d’acquisition du langage se situe entre 3 et 4 ans (retard non systématique).
Retrouvez des conseils, des infos et des outils sur les difficultés alimentaires de votre enfant sur le site ![]()
Prise en charge au sein des autres réseaux
Compte tenu des nombreuses manifestations cliniques, la prise en charge globale des patients atteints du syndrome de Costello nécessite une coopération pluridisciplinaire médicale, paramédicale et sociale. Pour la prise en charge des troubles ORL, des malformations faciales et des soins dentaires, le patient pourra être pris en charge au sein des autres réseaux TETECOU.
En savoir plus sur la prise en charge au sein des autres réseaux
Pour les atteintes intellectuelles, cardiaques, squelettiques (etc.) la prise en charge peut se faire au sein des filières santé maladies rares : Défiscience, AnDDI-Rares, Oscar et Cardiogen.
Pour aller plus loin
Association de patients
Association du syndrome de Costello et CFC
![]()
Recommandations et autres références pour les professionnels

Recommandation internationale
Costello syndrome: Clinical phenotype, genotype, and management guidelines
2019


Ressources Oprhanet
Page Orphanet
Syndrome de Costello

Page Orphanet
Article tout public
Syndrome de Costello

Ressources TETECOU
Page TETECOU
Syndrome de Costello

Podcast TETECOU
